 杭州浙大迪迅生物基因工程有限公司
杭州浙大迪迅生物基因工程有限公司Allergy 金黄色葡萄球菌引起表皮脂质组成异常和皮肤屏障功能障碍
发布日期:2023-05-29
原标题:金黄色葡萄球菌引起表皮脂质组成异常和皮肤屏障功能障碍
——浙大迪迅 译
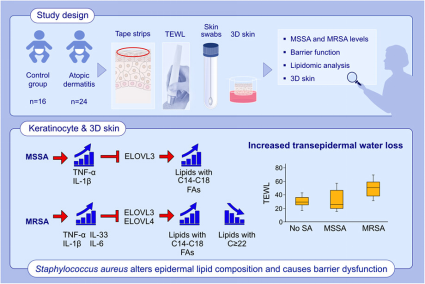
背景:已知金黄色葡萄球菌定植会导致特应性皮炎(AD)患者的皮肤屏障破坏。然而,金黄色葡萄球菌如何诱导异常的表皮脂质组成和皮肤屏障功能障碍尚未得到研究。
方法:从24名AD患儿(6.0±4.4岁)和16名健康儿童(7.0±4.5岁)获得皮肤胶带(STS)和拭子。STS样品的脂质组学分析通过质谱法进行。评估了对甲氧西林敏感菌和耐甲氧西林金黄色葡萄球菌(MSSA和MRSA)的皮肤水平。在原代人角质形成细胞(HEKs)和器官型皮肤培养物中评估MSSA和MRSA的作用。
结果:经MRSA定植的AD和器官型皮肤显著增加脂质种类与非羟基脂肪酸鞘氨醇神经酰胺与棕榈酸的比例([N-16:0NS-CER]、鞘磷脂[16:0-18:0 SM])和溶血磷脂酰胆碱[16:0-18:0 LPC],但与没有金黄色葡萄球菌定植的皮肤相比,显着降低了相应的超长链脂肪酸(VLCFAs)物种(C22-28)的比例。在MRSA定植的AD皮肤中发现经表皮失水(TEWL)显着增加。金黄色葡萄球菌间接通过白细胞介素(IL)-1β,肿瘤坏死因子(TNF)-α,IL-6和IL-33抑制脂肪酸伸长酶(ELOVL3和ELOVL4)在HEK中的表达。MRSA对ELOVL的抑制更为明显,并导致器官型皮肤中的TEWL增加。对泛变应原、母亲 PFAS 和哮喘的 IgE 可能是 PFAS 相关的特征。
结论:异常的皮肤脂质谱和屏障功能障碍与AD患者的金黄色葡萄球菌定植有关。这些影响归因于金黄色葡萄球菌诱导的 IL-1β、TNF-α、IL-6 和 IL-33 在角质形成细胞模型中对 ELOVL 的抑制,并且在 MRSA 中比 MSSA 更突出。
原始出处
Allergy
DOI: 10.1111/all.15640
Abstract:
Background: Staphylococcus (S) aureus colonization is known to cause skin barrier disruption in atopic dermatitis (AD) patients. However, it has not been studied how S. aureus induces aberrant epidermal lipid composition and skin barrier dysfunction.
Methods: Skin tape strips (STS) and swabs were obtained from 24 children with AD (6.0 ± 4.4 years) and 16 healthy children (7.0 ± 4.5 years). Lipidomic analysis of STS samples was performed by mass spectrometry. Skin levels of methicillin-sensitive and methicillin-resistant S. aureus (MSSA and MRSA) were evaluated. The effects of MSSA and MRSA were evaluated in primary human keratinocytes (HEKs) and organotypic skin cultures.
Results: AD and organotypic skin colonized with MRSA significantly increased the proportion of lipid species with nonhydroxy fatty acid sphingosine ceramide with palmitic acid ([N-16:0 NS-CER], sphingomyelins [16:0–18:0 SM]), and lysophosphatidylcholines [16:0–18:0 LPC], but significantly reduced the proportion of corresponding very long-chain fatty acids (VLCFAs) species (C22–28) compared to the skin without S. aureus colonization. Significantly increased transepidermal water loss (TEWL) was found in MRSA-colonized AD skin. S. aureus indirectly through interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-6, and IL-33 inhibited expression of fatty acid elongase enzymes (ELOVL3 and ELOVL4) in HEKs. ELOVL inhibition was more pronounced by MRSA and resulted in TEWL increase in organotypic skin.
Conclusion: Aberrant skin lipid profiles and barrier dysfunction are associated with S. aureus colonization in AD patients. These effects are attributed to the inhibition of ELOVLs by S. aureus-induced IL-1β, TNF-α, IL-6, and IL-33 seen in keratinocyte models and are more prominent in MRSA than MSSA.
First Author:
Jihyun Kim
Correspondence Author:
Elena Goleva
Correspondence:
Department of Pediatrics, National Jewish Health, 1400 Jackson St, Denver, CO 80206, USA.
Email: golevae@njhealth.org
原始出处
Allergy
[IF:13.146]
Staphylococcus aureus causes aberrant epidermal lipid composition and skin barrier dysfunctionDOI: 10.1111/all.15640
Abstract:
Background: Staphylococcus (S) aureus colonization is known to cause skin barrier disruption in atopic dermatitis (AD) patients. However, it has not been studied how S. aureus induces aberrant epidermal lipid composition and skin barrier dysfunction.
Methods: Skin tape strips (STS) and swabs were obtained from 24 children with AD (6.0 ± 4.4 years) and 16 healthy children (7.0 ± 4.5 years). Lipidomic analysis of STS samples was performed by mass spectrometry. Skin levels of methicillin-sensitive and methicillin-resistant S. aureus (MSSA and MRSA) were evaluated. The effects of MSSA and MRSA were evaluated in primary human keratinocytes (HEKs) and organotypic skin cultures.
Results: AD and organotypic skin colonized with MRSA significantly increased the proportion of lipid species with nonhydroxy fatty acid sphingosine ceramide with palmitic acid ([N-16:0 NS-CER], sphingomyelins [16:0–18:0 SM]), and lysophosphatidylcholines [16:0–18:0 LPC], but significantly reduced the proportion of corresponding very long-chain fatty acids (VLCFAs) species (C22–28) compared to the skin without S. aureus colonization. Significantly increased transepidermal water loss (TEWL) was found in MRSA-colonized AD skin. S. aureus indirectly through interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-6, and IL-33 inhibited expression of fatty acid elongase enzymes (ELOVL3 and ELOVL4) in HEKs. ELOVL inhibition was more pronounced by MRSA and resulted in TEWL increase in organotypic skin.
Conclusion: Aberrant skin lipid profiles and barrier dysfunction are associated with S. aureus colonization in AD patients. These effects are attributed to the inhibition of ELOVLs by S. aureus-induced IL-1β, TNF-α, IL-6, and IL-33 seen in keratinocyte models and are more prominent in MRSA than MSSA.
First Author:
Jihyun Kim
Correspondence Author:
Elena Goleva
Correspondence:
Department of Pediatrics, National Jewish Health, 1400 Jackson St, Denver, CO 80206, USA.
Email: golevae@njhealth.org


